

Biliary atresia is a liver condition in children wherein the bile fluids cannot flow from the hepatic tissues to the intestines, due to the bile ducts being rather narrow, obstructed or entirely missing. Like Alagille syndrome, biliary atresia is also a rare ailment, medically termed as extrahepatic ductopenia and progressive obliterative cholangiopathy. However, unlike the former, it is not inherited from either of the parents and is either a congenital defect present in the baby right from birth or acquired quite early on in childhood.
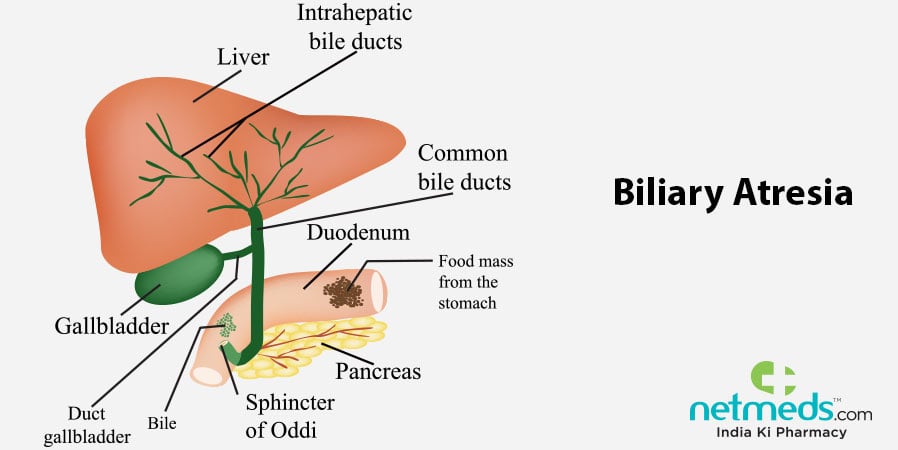
In a normal, healthy baby, the bile ducts are fully developed and properly aligned, with sufficient space within the tubules for passage of bile fluids. Bile is a liquid substance produced by the liver, which then travels via the bile ducts to the intestines to aid in digestion and subsequent assimilation of food particles. However, in instances of biliary atresia, owing to faulty or absent bile duct structures, bile cannot pass through to the intestines, thereby accumulating in the liver and resulting in fatty liver, haemochromatosis, hepatic cirrhosis and liver damage.
Also Read: Haemochromatosis: Causes, Symptoms And Treatment
Causes Of Biliary Atresia:
The exact cause of biliary atresia is not known, but medical experts ascertain that the contributing factors consist of:
- A hampered immune system in the newborn prompting inflammation and injury of hepatic tissues
- Genetic mutations that arise abruptly during foetal development or very early on in the newborn
- Coming in direct contact with toxic chemicals such as aflatoxin – a carcinogen and mutagen produced by fungi
- Viral infections like congenital cytomegalovirus (CMV) infection, that impair the formation of bile ducts
- Autoimmune reactions that hinder normal liver development
Risk Factors:
Certain aspects make some children more prone to acquiring biliary atresia than others, such as:
- Being female, since this liver disorder occurs more often in girls than boys
- Belonging to Asian or African American lineage, as more cases are reported among these populations than in Caucasians
- Developing diabetes during pregnancy i.e. gestational diabetes
Symptoms:
The characteristic indications of biliary atresia closely resemble those of newborn jaundice, including excess bilirubin in blood along with the yellowing of skin and eyes.
Additional symptoms of this liver condition comprise elimination of dark brown urine, the passage of oddly coloured beige stools, enlarged liver and spleen, challenges in gaining weight, fluid buildup in the stomach – ascites. If the child is not given appropriate medical treatment on time, biliary atresia eventually results in liver failure after many months.
Diagnosis:
The physician initially records the baby’s complete medical history and performs a thorough physical examination to look for any perceivable signs of liver problems such as a swollen area near the liver or spleen. The paediatrician also carries out a blood test to measure bilirubin levels in the blood and assess other parameters of liver function.
Furthermore, a portion of the liver tissue is excised, known as a liver biopsy, to study the sample for any abnormalities. The internal structures of the liver, bile ducts, gall bladder are all examined utilising ultrasound, hepatobiliary scan and cholangiogram procedures.
Treatment:
There exists no cure for biliary atresia in newborns and young children. Once the diagnosis of this hepatic defect is confirmed, the healthcare professional rectifies the liver structures by means of surgery.
A surgical technique called the Kasai procedure is performed in the affected child. It involves eliminating the impaired bile ducts outwardly from the liver and replacing them with a segment of the child’s small intestine. This ensures that the bile fluids can flow unobstructed from the liver directly into the small intestine.
In some cases, the surgeries are not successful or effective owing to complications in the patient. The child then requires a liver transplant to overcome biliary atresia and restore normal liver functions.
 Previous
Previous
