

Recently, a very powerful medication named Zolgensma was approved for use by the NHS i.e. National Health Service in the UK, two years after it was first permitted by the FDA (Food And Drug Administration) in the USA, in 2019. The medication was also officially sanctioned in Japan and the European Union a year ago, in 2020. This formulation, manufactured by a renowned American pharmaceutical company, Novartis, is currently the world’s most expensive drug, costing Rs. 18 crores for a single dose and is also made available in India. It is used as advanced gene therapy to treat a very rare genetic disease in children below 2 years, known as Spinal Muscular Atrophy (SMA). Gene therapy involves the precise introduction of genes i.e. sequences of DNA or nucleic acids into the cells in the body, to efficiently rectify genetic issues and resolve medical conditions.
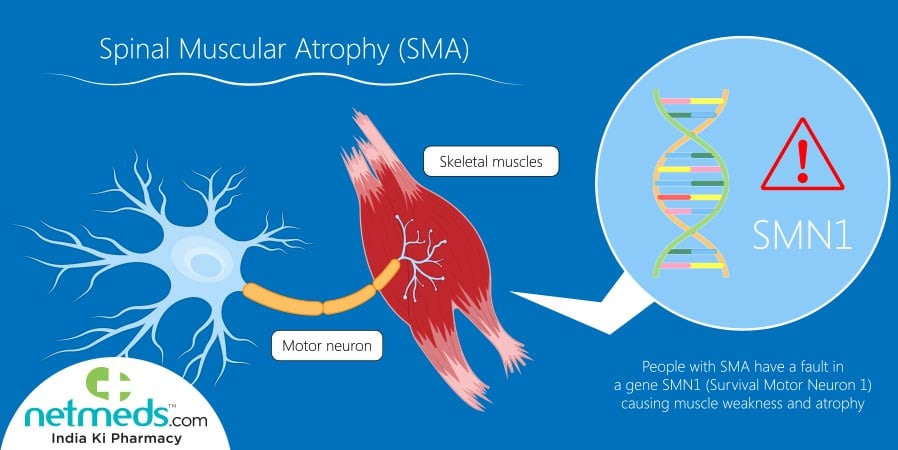
Spinal muscular atrophy is a debilitating illness characterised by a complete lack of control of muscle movements, due to damaged or absent nerve cells in the brain and spinal cord. It is a genetic ailment that mostly develops in children, like Alagille Syndrome, with those suffering from SMA surviving only up to 2 years of age. Hence the costly medication of Zolgensma significantly improves the grave symptoms and life expectancy of patients with SMA. Zolgensma being the brand name, the formal chemical and pharmaceutical terminology of this compound is onasemnogene abeparvovec. This medicine is administered intravenously i.e. through the vein as a one-time infusion and supplies a new replica of the gene that synthesizes the human SMN protein, otherwise denoted as SMN1 protein, Survival Motor Neuron 1 protein. Since this key protein controls the activity of muscles and glands in the system, a defect or deficiency in SMN1 results in spinal muscular atrophy.
It is thus important to gain in-depth insight into the types, causes, symptoms of spinal muscular atrophy, for accurate diagnosis, prompt treatment and extended lifespan of afflicted children.
Types Of Spinal Muscular Atrophy
There are 4 types of spinal muscular atrophy, depending on when the symptoms begin to display prominently and the life expectancy, quality of living standards in affected patients. These include:
SMA Type 1:
SMA Type 1, otherwise termed as Werdnig–Hoffmann disease that develops in infants less than 6 months old. Symptoms consist of a bent, arched spine, breathing difficulty, weak muscular tissues, inability to move and flex limbs and uncontrolled twitching, jerking of muscles. With timely medical treatment, this condition can be corrected, the lifetime of children can be extended up to young adulthood, with improved capacity to sit, stand and walk.
SMA Type 2:
This form of spinal muscular atrophy is also referred to as Dubowitz disease and occurs in children between the ages of 6 to 18 months. Affected children usually portray lung problems, shortness of breath and impaired muscle movement. Proper medical treatment helps the child to sit, but not stand or walk and raises the lifespan to survive until adolescence to young adulthood.
SMA Type 3:
SMA Type 3 or Kugelberg–Welander disease happens in children older than 18 months. Obvious signs comprise curved neck, spine or scoliosis, contracting of muscles, tendons and tremors, trembling sensations in fingers. Advanced medical treatment promotes the capacity to walk, but with abnormal positions while running, climbing stairs and bending forwards or backwards and appropriate timely healthcare also ensures normal life expectancy in affected children.
Also Read: Scoliosis: Causes, Symptoms And Treatment
SMA Type 4:
This is also known as adult-onset SMA, which occurs very rarely and in adults above the age of 21. SMA Type 4 induces weakness particularly in the proximal muscles i.e. the tissues adjacent and nearest to the centre of the body. However, it generally does not influence life expectancy, with normal lung capacity, breathing, respiratory wellness and the ability to sit, stand, walk freely, with only mild weakness in leg muscles.
Causes
Spinal muscular atrophy is triggered either from a lack of optimal operations of motor neurons in the brain stem and spinal cord, or genetic defects in a specific gene known as SMN1 i.e. survival motor neuron 1. Motor neurons being the specialised nerve cells that regulate the movement of muscles and functioning of glands in the body, genomic defects in SMN1 leads to instances of spinal muscular atrophy.
This is because the nerve signal from the brain or spinal cord to the muscles is not transmitted smoothly, being obstructed and resulting in a lack of coordination in muscle movement. SMA Types 1, 2 and 3 are usually inherited by the child only when both parents contain faulty SMN1 genes that are passed on to the offspring.
Symptoms
The symptoms of spinal muscular atrophy occur based on which type of SMA is present in the affected individual, as well as at what age the person obtains the genetic condition. The distinguishing indications of SMA comprise:
- Weakness in muscles with sudden jolting motions
- Difficulty in breathing and with swallowing food
- Irregular shape of limbs, chest, spine due to a decline in muscle strength
- Challenges with sitting, standing and walking normally
- Increased risk of acquiring respiratory problems and infections
Since spinal muscular atrophy is a degenerative neurological sickness, the symptoms tend to become exacerbated with time. However, proper and immediate medical treatment ensures improved life expectancy and better quality of living.
Diagnosis
When parents observe typical symptoms of SMA in their child, such as hindered muscle movement and breathing, swallowing difficulty and report it to the doctor at once, accurate diagnosis and timely effective treatment of SMA is possible.
The physician questions the parents about their complete family medical history and conducts a thorough physical exam of the affected child. This helps determine if the muscles are limp and tendon reflexes are hampered. The healthcare provider also performs blood tests, muscle biopsies and electromyography, to evaluate the functioning of motor neurons in the child.
Treatment
At present, there exists no cure for spinal muscular atrophy and no possible means to prevent the condition, since it is inherited in the child from both parents. Yet, proper advanced medical treatment significantly improves the life expectancy of the affected child and enhances the quality of living.
The medical specialist prescribes potent medications known as DMTs i.e. disease-modifying therapies and drugs that alter genetic mechanisms in the body, to alleviate symptoms of muscle weakness, convulsions, spinal posture and improve motor neuron function. Also, gene therapy in the form of Zolgensma is administered as an intravenous single-shot injection, to children less than 2 years old. This drug helps to rectify defects in SMN1 genes, promote motor neuron activity and improve life expectancy, living standards in kids diagnosed with SMA.
 Previous
Previous
